Hi Matt,
Thank you for trying out the Profile By Reference module. I have some solutions:
Spin systems slowing and crashing
Unfortunately, accurate spin-system simulations have an exponential complexity to generate. This means that moderately sized metabolites (>7 spins) require a large amount of time and memory to compute. We currently do not have an elegant solution to this, so for the time being, the simplest solution is to close any large simulated spin system spectra (using the cross on the simulation tab) before opening another. So long as you ‘commit’ the simulations to the profile record, using the ‘commit’ buttons, the sum-spectra will keep the shape of that metabolite, even after it is closed.
Closing and saving PBR
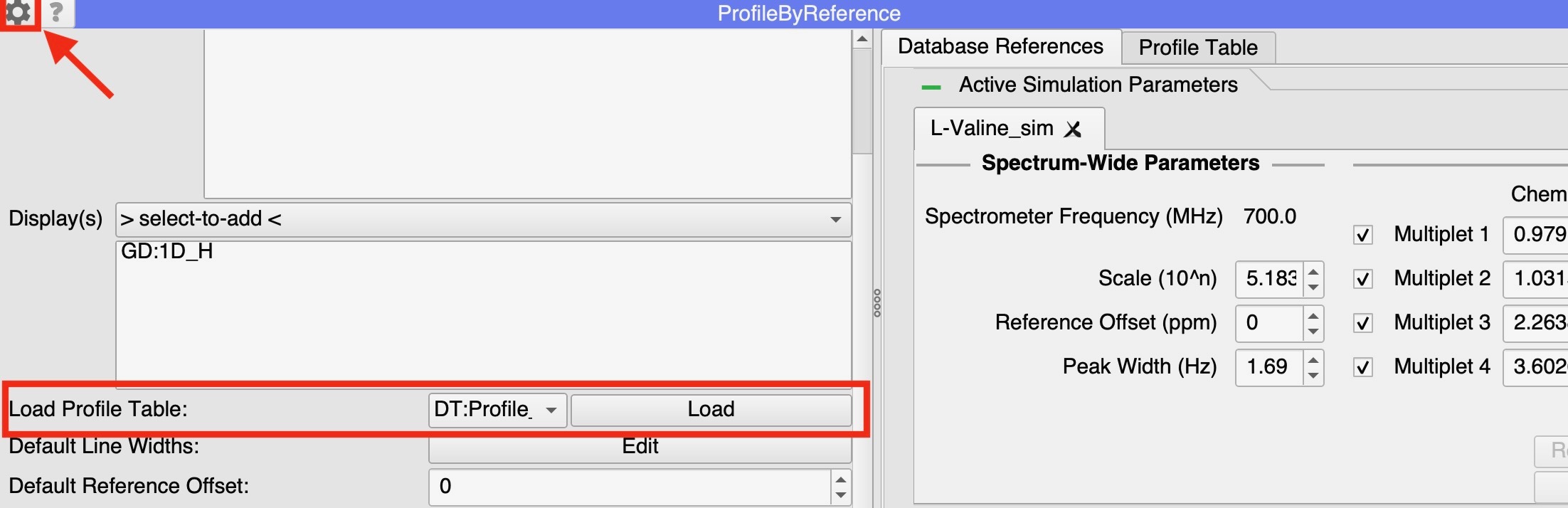
The PBR module holds on to all the simulated spectra data, so when it is closed, it is no longer possible to manipulate these spectra. However, all the data to recreate the state of the profile is stored in the ‘Profile Table’, which remains in the project as a ‘Data Table’. When opening a new PBR module, it is possible to load in the Profile Table from an existing Data Table:
- Open the settings menu in the PBR module (the cog button at the top left of the module)
- Scroll down to the pull-down menu beneath ‘Displays’ and select the table you want to load.
- Click ‘Load’
This may take a couple of minutes as it will have to re-create the Sum spectra. This new PBR module will not automatically return to the same state, so you will have to re-open any simulations to return them to their ‘active’ states. These can be conveniently found in the ‘Simulations in Profile’ table in the PBR module.
This approach was made primarily for sharing Profile Tables between projects, rather than reopening a closed module. Therefore the best way to keep the state of the profile is to keep the PBR module open until the profiling is complete. Saving a project will keep the state of the module and all active simulations, so you can close the program while PBR is open and it will return to the same state when you reopen the project. However, it will only remember the changes you have ‘committed’, so be sure to commit all changes to the profile you want to keep before closing the project.
I see that you have sometimes lost paths when you do this. I will investigate this.
Selecting and moving all peaks
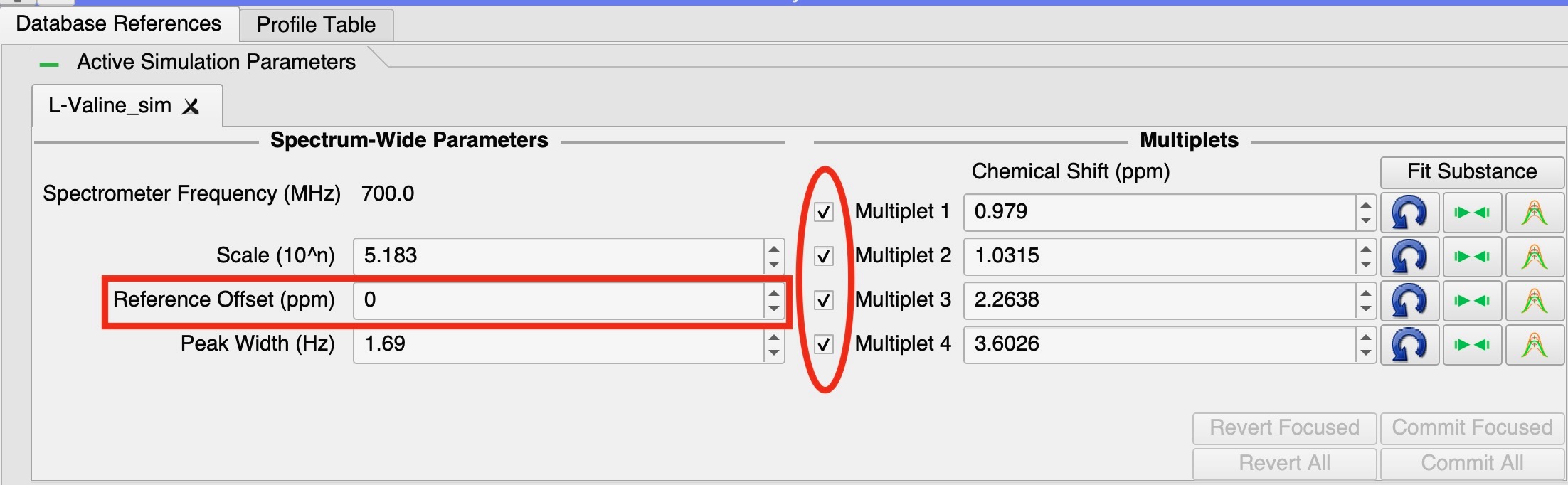
In the ‘Active Simulation Parameters’ frame in the PBR module. All simulations will have a spin-box labelled ‘Reference Offset (ppm)’. This spin-box moves the whole simulation by that value on the ppm axis, but it does not have a vertical-line to drag in the spectrum displays.
You can also multi-select the multiplets using the check-boxes to the left of the multiplet chemical-shift spin-boxes. All checked multiplets will move together whenever one of them is moved. Therefore by checking all of them, you can use any vertical-line for that simulation to drag all the peaks at once.
Phasing
The change of shape from phasing sounds like a bug in the 1D phasing console. We can investigate this.
Hopefully that helps a bit. There are also many updates to PBR that will be available soon, in advance of the upcoming CCPN Metabolomics workshop on 05/11/2024 in Liverpool. These include many robustness improvements, quality of life updates and out-fitting tools.
Let me know if you have any other questions or reports!
Thanks,
Morgan