Hi Max,
Copying the NmrChain would be quite straight forward: Go to in the sidebar and then select Clone from NmrChain:
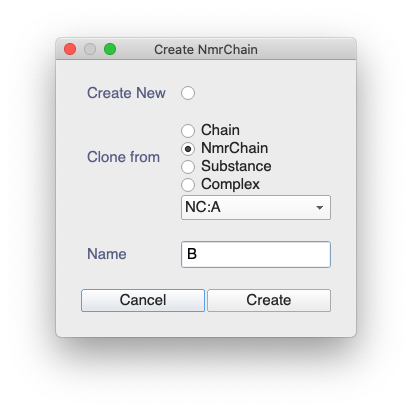
But that won’t automatically create the Chemical Shifts.
So instead I would suggest you run the following macro which should duplicate your chemical shifts:
csl = get('CL:default')
for cs in csl.chemicalShifts:
if cs.nmrAtom.nmrResidue.nmrChain.name != 'B':
naa = cs.nmrAtom
ncb = project.fetchNmrChain('B')
nrb = ncb.fetchNmrResidue(naa.nmrResidue.sequenceCode, naa.nmrResidue.residueType)
nab = nrb.fetchNmrAtom(naa.name)
csl.newChemicalShift(value = cs.value, valueError=cs.valueError, nmrAtom = nab)
Obviously you’ll need to make sure that your ChemShiftList at the top is the correct one and if you want to use anything other than ‘B’ as your chain name for the second chain, there are a couple of places you need to change that.
It may also be worth being aware that these ChemicalShifts will technically be ‘orphans’. Have a read of the text below (new documentation that isn’t online yet) to understand the way in which Version 3.1 handles Chemical Shifts and what orphans are (you may be aware from V2 already).
When you create a NEF file you can choose to include or exclude orphans. In this case you obviously need to include them. However, be aware that this could include some other rogue orphans that you already have in your chemical shift list. It could be worth making sure you delete those before you start duplicating the chemical shifts. (Probably best to save a backup of your project before you start all of this!).
This will obviously put your B Chemical Shifts in the same ChemShiftList as your A ChemShifts. If you want them in a different one, then simply create a new ChemShiftList and change the code to:
csl = get('CL:entry17285')
cslb = get('CL:BChemShifts')
for cs in csl.chemicalShifts:
if cs.nmrAtom.nmrResidue.nmrChain.name != 'B':
naa = cs.nmrAtom
ncb = project.fetchNmrChain('B')
nrb = ncb.fetchNmrResidue(naa.nmrResidue.sequenceCode, naa.nmrResidue.residueType)
nab = nrb.fetchNmrAtom(naa.name)
cslb.newChemicalShift(value = cs.value, valueError=cs.valueError, nmrAtom = nab)
In this case the B ChemShifts will be static rather than orphans.
Best wishes,
Vicky
Chemical Shifts
Note that this is new for Version 3.1 - in Version 3.0 the Chemical Shifts are handled slightly differently
Each project always has to contain at least one Chemical Shift List. New projects are given one called default.
The link between Spectra and Chemical Shift Lists
Each spectrum has to be associated with one Chemical Shift List. When you open a new spectrum, it will by default be associated with the first Chemical Shift List in your project (usually the default list). If you double-click on a spectrum in the sidebar to bring up the Spectrum Properties dialog box, you can see in the General tab which Chemical Shift List the Spectrum is associated with. You can also change which Chemical Shift List this is (including creating a new Chemical Shift List) using the drop-down menu.
An alternative way to manipulate the link between a Spectrum and a Chemical Shift List, is to double-click on a Chemical Shift List in the sidebar. This will show you which spectra are associated with this Chemical Shift List and which are not. You can drag spectra between the Not Included and Included boxes to change which spectra belong to that Chemical Shift List (or double-click on a spectrum to move it to the other box). If you only have one Chemical Shift List in your project, you will not be able to exclude spectra from this. If you move spectra out of the current Chemical Shift List, then they will by default go into the top or next available Chemical Shift List.
When you assign peaks in a spectrum, then the NmrAtoms used for the assignment will be added to the Chemical Shift List of that spectrum. The Chemical Shift value for any one NmrAtom will then be automatically averaged over all the Peaks it has been assigned to in Spectra which are linked to that Chemical Shift List. We refer to these Chemical Shifts as being dynamic because they change dynamically as you assign and deassign peaks in the linked spectra.
If a Chemical Shift for a particular NmrAtom was added to a Chemical Shift List, but later the assignment is removed (i.e. the NmrAtom is no longer being used for any peak assignments), then the Chemical Shift remains in the list, but it is marked as being orphaned because it no longer has a link to any peaks. It’s Chemical Shift value is simply the last value it had. Note that when you export a NEF file from your project you can choose whether or not to include ChemicalShift orphans. Usually you will not want to export these as they are often simply the remnants of incorrect or temporary assignments.
When should I use multiple Chemical Shift Lists?
You should use additional Chemical Shift Lists any time you add spectra to your project which are recorded under different conditions resulting in (potentially) changed chemical shifts. So if you record some spectra at a different temperature, a different pH or including different concentrations of a ligand or binding partner, then you should use a different Chemical Shift List. So for a titration, each of your spectra should (ideally) be in a separate Chemical Shift List. Otherwise, your Chemical shifts will be average across all the spectra in the titration. If you did not put them in different Chemical Shift Lists, the chemical shifts of the peaks that move will contribute towards the chemical shift of that NmrAtom. And when you go to deposit your Chemical Shift List, some of your chemical shifts might be distorted!
Duplicating and Importing Chemical Shift Lists
You can duplicate a Chemical Shift List by right-clicking on it in the sidebar and selecting Duplicate. This will give you a snap-shot of your Chemical Shift List at that point in time and because the List isn’t associated with any spectra, the Chemical Shifts in it won’t change as you manipulate the peaks in your spectra. We refer to the chemical shifts in such a list (which doesn’t have any spectra associated with it) as being static because they won’t change.
In a similar way, you can import a Chemical Shift List from a BMRB NMRStar or NEF file and then that Chemical Shift List will also not be associated with any spectra and have static Chemical Shifts.
If you later associate a spectrum with a Chemical Shift List of this type, then chemical shifts all become dynamic as they can now be automatically altered through peak assignments and deassignments.
Dynamic, Static and Orphaned Chemical Shifts
Dynamic Chemical Shifts are in a Chemical Shift List which is linked to at least one spectrum. Static Chemical Shifts are in a Chemical Shift List which is not linked to any spectra. A dynamic chemical shift is orphaned if its NmrAtom is not used for any peak assignments in the linked spectra.
If you would like to see whether your Chemical Shifts are dynamic or static or if any of your dynamic Chemical Shifts are orphaned, you can open a Chemical Shift Table (drag it into the Drop Area from the sidebar, or right-click in the sidebar and select Open as Module) and then right-click on a column heading and select Column Settings… You can then select to view the State and Orphaned columns to see if your Chemical Shifts are dynamic or static. A tick in the Orphaned column indicates that a dynamic Chemical Shift is orphaned.