Hi,
I was trying to analyze the analyze the titration data. I have imported the sparky project to ccpnmr, but different .save files have different chain names. Is there any way to make the same chain name for all these Spectra.
Thank you
(04-14-2020, 01:43 pm)ashubiotech Wrote: Hi,
I was trying to analyze the analyze the titration data. I have imported the sparky project to ccpnmr, but different .save files have different chain names. Is there any way to make the same chain name for all these Spectra.
Thank you
Hello,
One option might be to simply edit the .save files? They are simple text files so you maybe can easily do a search/replace on them, to make the assignments (more) consistent?
Best wishes,
Patrick
Hi,
hopefully the following macro should move all your peak assignments into a single NmrChain called `molecule` (you can re-name it afterwards) and then remove all the other NmrChains created during import.
Python
- # Do this on every peak in the project
- for peak in project.peaks:
- # Get the assignments in all dimensions for a peak
- for assignOptions in peak.assignedNmrAtoms:
- for assignment in assignOptions:
- # Create the new assignment string with `molecule` as the NmrChain
- temp = str(assignment)
- assignmentComponents = temp.split(`.`)
- assignmentComponents[0] = `NA:molecule`
- assignmentComponents[3] = assignmentComponents[3][0:-1]
- newAssignment = `.`.join(assignmentComponents)
- # get parameters needed for creation of new NmrChain, NmrResidue, NmrAtom and Peak Assignment
- chainPid = `NC:molecule`
- resPid = `NR:molecule.` + `.`.join(assignmentComponents[1:-1])
- seqCode = assignmentComponents[1]
- resType = assignmentComponents[2]
- atomName = assignmentComponents[3]
- axCde = assignmentComponents[3][0]
- # Create new NmrChain, NmrResidue and NmrAtom if necessary
- project.fetchNmrChain(`molecule`)
- get(chainPid).fetchNmrResidue(seqCode, resType)
- get(resPid).fetchNmrAtom(atomName)
- # Change the peak assignment to the new one
- get(peak.pid).assignDimension(axCde, newAssignment)
-
- # Remove all the (now obsolete) NmrChains that are not NC:molecule and the default NC:@-
- for nmrChain in project.nmrChains:
- if nmrChain.id != `@-`:
- if nmrChain.id != `molecule`:
- project.deleteObjects(nmrChain)
If you need to switch your amino acids from one-letter to three-letter code you can use the following:
Python
- def convertResidueCode(residueName, inputCodeType=`oneLetter`, outputCodeType=`threeLetter`, molType =`protein`):
- """
- :param inputCodeType: oneLetter, threeLetter, synonym, molFormula
- :type inputCodeType: str
- :param molType: `protein`, `DNA`, `RNA`
- :type molType: str
- :return: the same residue with the new letter code/name
- :rtype: str
- """
- from ccpnmodel.ccpncore.lib.chemComp.ChemCompOverview import chemCompStdDict
- modes = [`oneLetter`, `threeLetter`, `synonym`, `molFormula`] # order as they come from ChemCom dictionary
- if inputCodeType not in modes or outputCodeType not in modes:
- print(`Code type not recognised. It has to be one of: `, modes)
- return
- for k, v in chemCompStdDict.get(molType).items():
- dd = {i:j for i,j in zip(modes,v)}
- if residueName == dd.get(inputCodeType):
- return dd.get(outputCodeType)
-
-
- def convertNmrChain1to3LetterCode(nmrChain, molType=`protein`):
- """
- converts NmrResidues from 1 to 3 LetterCode.
- :param nmrChain: Ccpn object NmrChain
- :param molType: `protein`, `DNA`, `RNA`
- :return:
- """
- for nmrResidue in nmrChain.nmrResidues:
- if len(nmrResidue.residueType) == 1:
- newNmrResidueName = convertResidueCode(nmrResidue.residueType, inputCodeType=`oneLetter`, outputCodeType=`threeLetter`, molType=molType)
- try:
- nmrResidue.rename(`.`.join([nmrResidue.sequenceCode, newNmrResidueName]))
- except Exception as err:
- print(`Error renaming NmrResidue %s.` %nmrResidue.pid, err)
- else:
- if not nmrResidue.residueType:
- print(`Skipping... ResidueType not found for nmrResidue %s` %nmrResidue.pid)
- else:
- print(`Skipping... Could not rename to 3 Letter code for nmrResidue %s` % nmrResidue.pid)
-
-
- # Do this on every NmrChain in the project
- for nmrChain in project.nmrChains:
- convertNmrChain1to3LetterCode(nmrChain)
Best wishes,
Vicky
Thanks Vicky,
it worked perfectly. I was able to get the binding curve but the window for fitting was empty. Is there any mistake? Another thing is I could not change the relative contribution of particular atoms (nitrogen and proton).
Ashish
Hi Ashish,
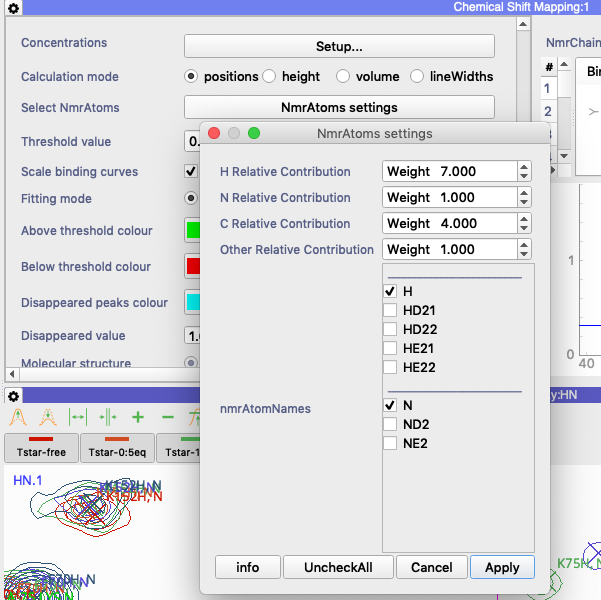
in the Chemical Shift Mapping module, try going to the settings (click on gear icon in the top left corner). Then scroll down to `Select NmrAtoms` and click on the `NmrAtoms settings` box (see attached figure). Here you can change the contributions for each of the different isotope types.
I`m not quite sure why the Fittings window is empty - though it is for me as well, so this is probably a bug of some kind. Will investigate.
Best wishes,
Vicky
Hi Ashish,
I`ve had another play and it looks as though the fitting should show once you click on a new residue in the table or graph. For some residues you might not see a fit, e.g. if the data is really bad because a peak doesn`t move as part of the titration.
Best wishes,
Vicky
Thanks Vicky,
Although I had tried to see the fitting curve but did not get successful. I got only the binding curves, not actual fitting.
Ashish