First and foremost, I want to say I’m really enjoying the 3.2 version and love it so far, thank you for all the efforts! Last week when Geerten came visiting Toronto, we had a brief discussion on some potential ssNMR related implementations in CCPN (I think one of the topics was brought up in a previous post recently). I will try summarizing the problems and potential improvements that may be considered in the next update.
Very often we want to display a peak list and put all the possible peak labels on certain spectrum to check things such as peak shifts upon protein interactions. I know that this can be done by generating a “simulated spectrum” of that certain type and the corresponding peak list after importing BMRB str file or a chemical shift table. However, the type of “simulated spectrum” and peak list that can be generated is only limited to some solution NMR type such as H/N HSQC and we can’t even generate basic C/C spectrum.
I see that this had been brought up in a previous post, and hopefully we could have all ssNMR types available in the future: 2D C/C, 2D N/CA and N/CO-1, 3D CA/N/CO-1, 3D N/CA/CX and N/CO-1/CX-1. (we are not quite sure what you meant by “the relayed version and only get the intra-residue CA-CX or CO-CX peaks”. Does it mean it can only display the C/C part in a NCACX or NCOCX spectrum?)
My CS assignment strategy starts with picking and labelling all the peaks in the CANCO spectrum. After finishing automatic peak picking and “Set Up NmrResidues”, the program only recognises the three atoms of the peaks in this spectrum as C-N-C, and I have to go to NmrAtom Assigner to manually change the first to CA[i] and the last to CO[i-1] (this also generates the corresponding @X-1 system for every @X residue, which is what I need for the backbone walk), for over 100-200 residues. Although it is possible to do this manually, repeating over 100+ residues is a tedious task. I’ve tried to set up the experiment type and the corresponding atom type for each dimension in the spectrum properties, but it doesn’t seem to do anything.
Is it possible to implement the ability in “Set Up NmrResidues” to automatically recognise and set up the NmrAtoms based on the atom type I select for each dimension? (In this case, I know exactly my atoms should be CA[i]/N[i]/CO[i-1] already.)
When I’m doing the backbone walk and connecting NmrResidues, sometimes I come across ambiguities in the CX-1 strip, e.g. there are two strong potential CA-1 peaks, or there are peaks that could be either CB-1 or a Glycine CA-1. I would like to explore all the possible options when facing ambiguities, by duplicating the NmrChain (containing the segments I connected already, e.g. chain NC:#2, up to the ambiguous NmrResidue), keep connecting through one option in the duplicated NmrChain and see where this leads – if I come across another ambiguity or realise this new connection through the duplicated chain may be a mistake, I could just go back the original chain segment before the duplication and attempted another connection, easily starting over from another option where I “branched out” (and delete this duplication one later if I can confirm that this path is actually wrong; right now I have to write them down on a piece of paper, and have to remember where I “branched out” options).
I’ve tried to “clone” a NmrChain (segment) in the Create New NmrChain option, but it always gives me an error message “Cannot create NmrResidue with reserved name @XX”.And If there is a way to simply move some NmrResdiues in one NmrChain to another NmrChain, that will be great too.
I’ve added all of these in now and will let you know once they are on the update server ready for download.
What I meant with the “relayed version” is basically that it’s the equivalent of using a short mixing time for the CX transfer, so you only get intra-residue links. We do ultimately want to add the ability to specify either the number of i+/-residues to transfer to or a distance cut-off based on a structure. These would give you the equivalent of longer mixing time experiments, but that will take a bit longer for us to implement.
I’ve written a macro which will do this for now. I’ll pop it in a separate post. At the moment it’s very much command line and will need a small amount of explaining. I’ll turn it into a more user-friendly pop-based one in due course. And ultimately, we can hopefully incorporate it into the main Set Up NmrResidues pop-up.
Geerten mentioned this and we discussed it in our group meeting yesterday. At the moment the program is limiting the creation of NmrResidues starting with @. But we have come to the conclusion that this is somewhat historical and that there is no real need to be so restrictive, so we are certainly planning to make this easier/possible.
Here is the macro which will set up the NmrResidues in any NMR experiment, giving you control over the atom names and also the offsets that you want to use.
You’ll see that at the top of the macro there are five variables for you to set. peakList: enter the the PID of the peakList you want to use nmrChain: enter the PID of the nmrChain you want to use keepAssignments: set to True of False depending on your preference nmrAtomNames: these are the names that will be used for each of the dimensions of your experiment. The order should correlate with the order in the Dimensions tab of the Spectrum Properties pop-up (double-click on the Spectrum in the sidebar). offsets: These are the relative offsets of the atoms, enter either 0 or -1. The order needs to correspond to that of the nmrAtomNames
If you are using a 2D experiment (e.g. NCO), then only enter two values for the nmrAtomNames and offsets, e.g. [‘C’,‘N’] and [’-1’,‘0’].
from ccpn.core.lib.ContextManagers import undoBlockWithSideBar as undoBlock
from ccpn.ui.gui.widgets.MessageDialog import showWarning
peakList = get('PL:CANCO.1')
nmrChain = get('NC:@-')
keepAssignments = True
nmrAtomNames = ['C','N','CA']
offsets = ['0', '0', '0']
def fetchNewPeakAssignments(peakList, nmrChain, keepAssignments, nmrAtomNames, offsets):
offsetsOkay = True
for offset in offsets:
if offset !='0' and offset != '-1':
offsetsOkay = False
if offsetsOkay is True:
if peakList.peaks:
peak = peakList.peaks[0]
nameIsoOffset = list(zip(nmrAtomNames, peak.spectrum.isotopeCodes, offsets))
with undoBlock():
for cc, peak in enumerate(peakList.peaks):
# only process those that are empty OR those not empty when checkbox cleared
dimensionNmrAtoms = peak.dimensionNmrAtoms # for speed reasons !?
if not keepAssignments or not any(dimensionNmrAtoms):
# make a new nmrResidue with new nmrAtoms and assign to the peak
nmrResidue = nmrChain.newNmrResidue()
if '-1' in offsets:
nmrResidueM1 = nmrChain.fetchNmrResidue(nmrResidue.sequenceCode + '-1')
nmrReses = []
for off in offsets:
if off == '0':
nmrReses.append(nmrResidue)
elif off == '-1':
nmrReses.append(nmrResidueM1)
nameIsoRes = list(zip(nmrAtomNames, peak.spectrum.isotopeCodes, nmrReses))
newNmrs = [[res.fetchNmrAtom(name=name, isotopeCode=isotope)] for
name, isotope, res in nameIsoRes]
else:
newNmrs = [[nmrResidue.fetchNmrAtom(name=name, isotopeCode=isotope)] for
name, isotope, offset in nameIsoOffset]
peak.assignDimensions(axisCodes=peak.axisCodes, values=newNmrs)
else:
showWarning('Wrong offset selected', 'Please make sure your offsets are only 0 or -1.')
fetchNewPeakAssignments(peakList, nmrChain, keepAssignments, nmrAtomNames, offsets)
I’ve tested the CANCO-1 NMRresidue setup amarcos, it works great.
In the new update, I’ve tested simulating all the ssNMR spectra, they all worked well except for the 2D C/C. There are two minor problems I hope you could help with:
The first one is minor, when I want to generate the simulated C/C, it seems all the available carbon types are simulated and this took a VERY long time and lots of resouces - I thought my computer was frozen for multiple times. On my laptop, it takes 15-30min to generate the peaklist and on my desktop, it still takes minutes to tens of minutes. On top of that, copying the position and assignment from the peaklist to my own spectrum also takes a very long time (especially when selected “snap to extremum” and “refit/recalculate peaks”. I think this is due to enormous amount of C/C peaks/coordinates - I do see there is an advance option when simulate the peaklist, but it doesn’t allow to control which carbons to be included. I wonder if there is a way to speed up the algorithm, or give the choice to select which carbons to be included (sometimes I don’t need to all the side chain carbons as I don’t see them anyway).
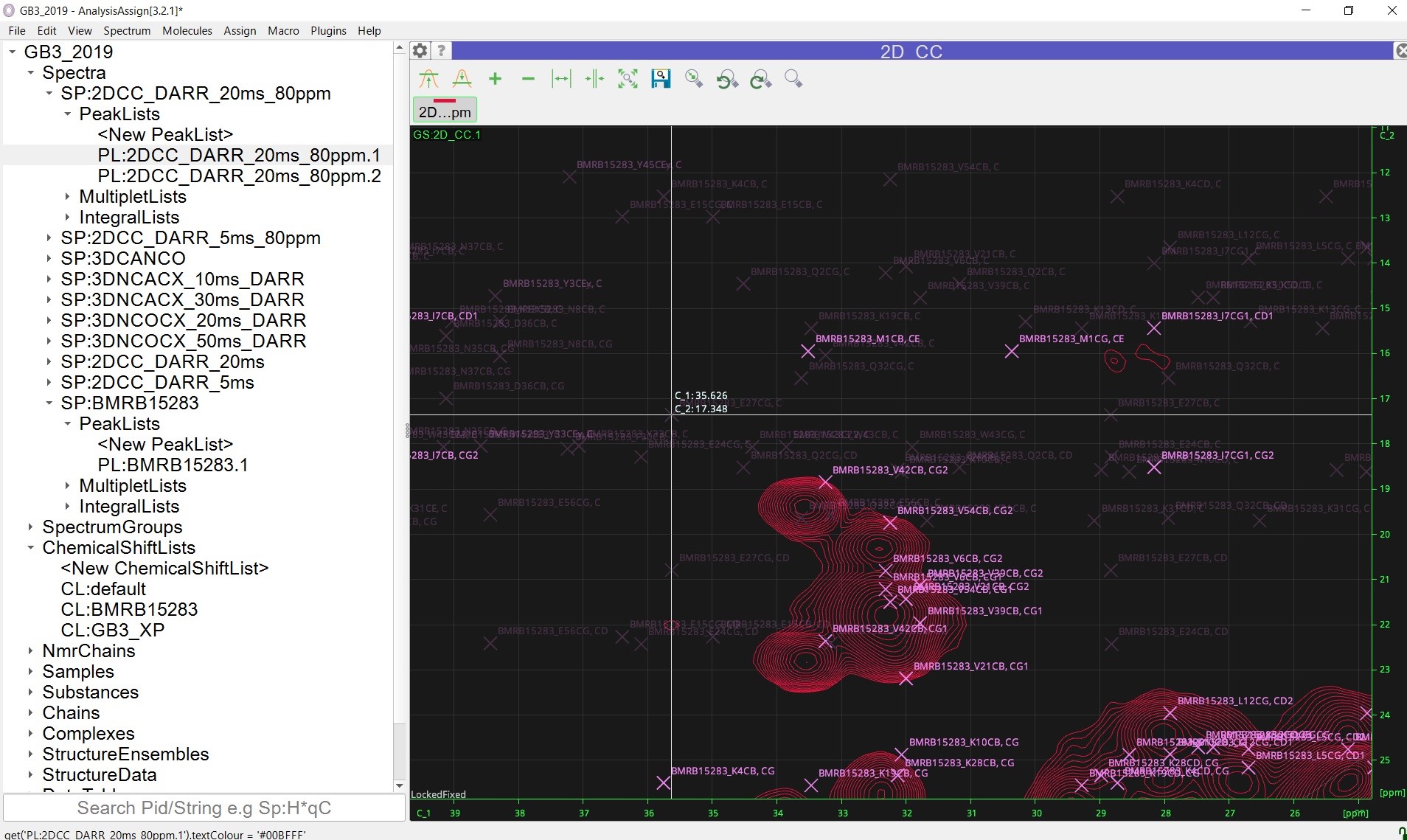
Another problem is that, when I have a “cropped” spectrum, i.e. cut the spectrum to certain region during data processing, the simulated peaklist will display on the wrong spots, probably due to spectrum fold. Here I included a screenshot of an example, I have a C/C spectrum with about 300 ppm original spectrum width, and I cut to only process the 0-80 ppm region while processing it in NMRpipe. The loaded simulated peaklist in this “cropped” spectrum looks like this:
Is there a way to fix this problem? The peak display won’t have such problem when I use the spectrum with the whole spectrum processed (but then, phasing the entire carbon spectrum is hard, rather than just a smaller region).
I forgot to mention, there is another minor thing, when I try to drag a NMRresidue in a NMRchain onto a NCO-1 spectrum to display the coordinate/crosshair of this N/CO-1 peak, it doesn’t know it should be the CO-1 one for the carbon dimension, only uses the CO of its own residue.
I’m aware that the algorithm is very inefficient, though we won’t be fixing this until we completely overhaul the simulated Peak List creation.
But I do have an old macro for making peaks in CC spectra which is a bit more efficient. I’ve added a couple of lines to removes any of the aliased peaks (i.e. ones that are out of bounds of the spectrum if you have cropped it).
I’ve also added some code which would allow you to restrict the peaks to particular atomNames if you wish (uncomment lines 13 and 22 and amend the atomList as appropriate. Obviously change the NmrChain and peakList PIDs near the top of the macro. One massive proviso (and part of the reason I didn’t mention this macro before, is that it will simply use the first ChemicalShiftList for the NmrAtoms in question). I would need to modify the macro further so you can specify the ChemicalShiftList of your choice. Will try and do that tomorrow.
Here is the code:
#Macro to create a CC peak list from a chemical shift list
from ccpn.core.lib.ContextManagers import undoBlock
# specify NmrChain associated with the imported chemical shifts
nmrChain = get('NC:A')
# specify CC PeakList to place the peak into
peakList = get('PL:sh3_uni_pdsd100.1')
# find carbon dimensions
carbonAxes = [axis for axis in peakList.spectrum.axisCodes if axis.startswith('C')]
# List of atoms to use (optional - uncomment lines 13 and 22 if you want to use this feature)
# atomList = ['CA', 'CB', 'C']
def makePairs(theList):
"make pairs from the elements of theList (excluding same-item pairs); return a list of tuples"
result = []
for idx, item1 in enumerate(theList):
for item2 in theList[idx+1:]:
# uncomment lines 13 and 22 if you want to use atom pairs from the atomList only
# if item1.name in atomList and item2.name in atomList:
result.append( (item1, item2) )
result.append( (item2, item1) )
return result
with undoBlock():
# Loop through each NmrResidue in the NmrChain
for nmrRes in nmrChain.nmrResidues:
# Loop through each NmrAtom, selecting each carbon
naList = [nmrAtom for nmrAtom in nmrRes.nmrAtoms if nmrAtom.name.startswith('C')]
# create all combinations of C-C peaks
pairs = makePairs(naList)
for na1, na2 in pairs:
peak = peakList.newPeak(ppmPositions=(na1.chemicalShifts[0].value, na2.chemicalShifts[0].value))
if peak.aliasing != (0,0):
peak.delete()
else:
peak.assignDimension(carbonAxes[0], na1)
peak.assignDimension(carbonAxes[1], na2)
peak.height = peakList.spectrum.getHeight((na1.chemicalShifts[0].value, na2.chemicalShifts[0].value))
I tested the macro and it works pretty well. Here it first fetches the NmrChain which is supposed to be associated with a chemical shift list - I wonder how are they associated? Right now it is like you said, it just uses the first ChemicalShiftList and the NmrChain name and the ChemicalShiftList name don’t need to be the same.
When you have time, could you also make some adjustment for dragging a NMRresidue in a NMRchain onto a spectrum to display the crosshair of the corresponding peak based on the spectrum type - right now this doesn’t recognize CO-1 for the carbon dimension, and only uses the CO of its own residue in a NCO-1 spectrum.
I forgot to mention, there is another minor thing, when I try to drag a NMRresidue in a NMRchain onto a NCO-1 spectrum to display the coordinate/crosshair of this N/CO-1 peak, it doesn’t know it should be the CO-1 one for the carbon dimension, only uses the CO of its own residue.
Is there a way to adjust this?
Sorry I didn’t have time to get back to you on this one yesterday.
I completely see why you would want this feature, but I’m afraid it’s probably not going to be possible any time soon.
One issue is that it means using the experiment types to determine which marks to draw - we don’t currently have any code that does this sort of thing. Once we sort out the synthetic peak lists properly, it should become much easier to implement something like this, though.
The other issue, however, is that it would be a change in philosophy. At the moment it is very straight forward: if you drag an NmrResidue, you get the marks for all the NmrAtoms in that NmrResidue (I think at the ChemicalShifts for the spectra in the SpectrumDisplay). Filtering and adjusting these by ExperimentType and then showing some i-1 or i+1 residue NmrAtom lines is a rather different proposition. And for experiments like the NCO, the question would be whether to show
the dragged residue N and i-1 CO
or
the dragged reside CO and the i+1 N
For some of the solution-style 3Ds with H(i)-N(i)-Ca/b(i-1) type peaks I think it would be even harder to determine which way round you would want to show the marks (and in some cases the pulse sequences can be either way round, so you can’t easily say “do it in the order in which the pulse sequence/coherence transfers are done”).
So I think the main barrier to implementing this feature is that I’m not sure if it would be possible to come up with a system which would be consistent and self-evident to users.
Here is new implementation of the macro which will work off a chemicalShiftList rather than an NmrChain. The algorithm probably looks rather convoluted, but is in fact considerably quicker than a conceptually easier NxN algorithm that loops only over the ChemicalShiftList.
#Macro to create a CC peak list from a chemical shift list
from ccpn.core.lib.ContextManagers import undoBlock
# specify ChemicalShiftList from which to generate Peaks
csl = get('CL:bmrb7106')
# specify CC PeakList to place the peaks into
peakList = get('PL:sh3_uni_pdsd100.1')
# List of atoms to use (optional - uncomment lines 12 and 24 if you want to use this feature)
# atomList = ['CA', 'CB', 'C']
# find carbon dimensions
carbonAxes = [axis for axis in peakList.spectrum.axisCodes if axis.startswith('C')]
def makePairs(theList):
"make pairs from the elements of theList (excluding same-item pairs); return a list of tuples"
result = []
for idx, item1 in enumerate(theList):
for item2 in theList[idx+1:]:
# uncomment lines 12 and 24 if you want to use atom pairs from the atomList only
# if item1.name in atomList and item2.name in atomList:
result.append( (item1, item2) )
result.append( (item2, item1) )
return result
with undoBlock():
# Find NmrResidues in CSL
nmrResidueList = []
for cs in csl.chemicalShifts:
if cs.nmrAtom.nmrResidue not in nmrResidueList:
nmrResidueList.append(cs.nmrAtom.nmrResidue)
# Loop through each NmrResidue in the CSL
for nmrRes in nmrResidueList:
# Loop through nmrResidue's NmrAtoms, selecting only the carbons
naList = [nmrAtom for nmrAtom in nmrRes.nmrAtoms if nmrAtom.name.startswith('C')]
# create all combinations of C-C peaks
pairs = makePairs(naList)
for na1, na2 in pairs:
for cs1 in na1.chemicalShifts:
if cs1.chemicalShiftList == csl and peakList.spectrum.aliasingLimits[0][0] < cs1.value < peakList.spectrum.aliasingLimits[0][1]:
for cs2 in na2.chemicalShifts:
if cs2.chemicalShiftList == csl and peakList.spectrum.aliasingLimits[1][0] < cs2.value < peakList.spectrum.aliasingLimits[1][1]:
peak = peakList.newPeak(ppmPositions=(cs1.value, cs2.value))
peak.assignDimension(carbonAxes[0], na1)
peak.assignDimension(carbonAxes[1], na2)
peak.height = peakList.spectrum.getHeight((cs1.value, cs2.value))