Hello Vicky, Thank you for the wonderful update!
I’ve tested the CANCO-1 NMRresidue setup amarcos, it works great.
In the new update, I’ve tested simulating all the ssNMR spectra, they all worked well except for the 2D C/C. There are two minor problems I hope you could help with:
The first one is minor, when I want to generate the simulated C/C, it seems all the available carbon types are simulated and this took a VERY long time and lots of resouces - I thought my computer was frozen for multiple times. On my laptop, it takes 15-30min to generate the peaklist and on my desktop, it still takes minutes to tens of minutes. On top of that, copying the position and assignment from the peaklist to my own spectrum also takes a very long time (especially when selected “snap to extremum” and “refit/recalculate peaks”. I think this is due to enormous amount of C/C peaks/coordinates - I do see there is an advance option when simulate the peaklist, but it doesn’t allow to control which carbons to be included. I wonder if there is a way to speed up the algorithm, or give the choice to select which carbons to be included (sometimes I don’t need to all the side chain carbons as I don’t see them anyway).
Another problem is that, when I have a “cropped” spectrum, i.e. cut the spectrum to certain region during data processing, the simulated peaklist will display on the wrong spots, probably due to spectrum fold. Here I included a screenshot of an example, I have a C/C spectrum with about 300 ppm original spectrum width, and I cut to only process the 0-80 ppm region while processing it in NMRpipe. The loaded simulated peaklist in this “cropped” spectrum looks like this:
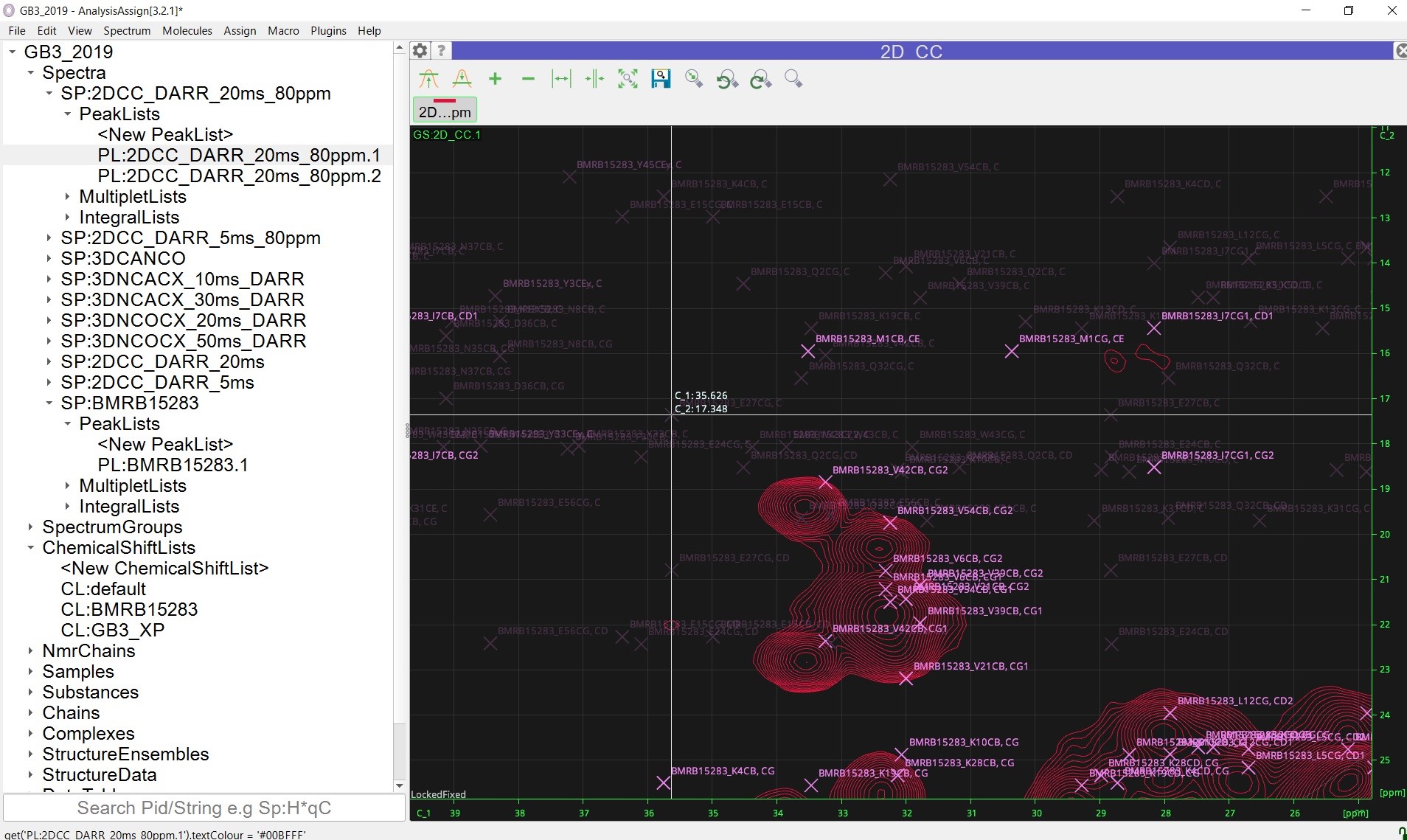
Is there a way to fix this problem? The peak display won’t have such problem when I use the spectrum with the whole spectrum processed (but then, phasing the entire carbon spectrum is hard, rather than just a smaller region).