I am working on a ligand-protein complex. The ~117 residue protein binds a sterol. We have a model of the protein structure from an ARTINA/NMRtist run and I have imported the assigned 13C-/15N edited NOESY peaks into Analysis. I have also imported the ligand as a smile string to create a 2nd NmrChain B (see screenshot). I have a 3D w1-13C half-filtered, w2-13C-/15N-edietd NOESY and a w1-,w2-double 13C half-filtered NOESY (peaks are too broad in the TOCSY to be much use, due to exchange [?]). How do I set-up the project (peaklists) so that CCPN know that the signals in the 2D NOESY of the sterold bound to the protein come from the ligand resonances, and how do I tell CCPN that in the 3D w1 half-filted, w2-13C-edited NOESY that the signals in w1 are ligand resonances and the signals in w2 & w3 are protein resonances?
Thanks,
Mark
Hi Mark,
At the moment that is no really way to do it unfortunately.
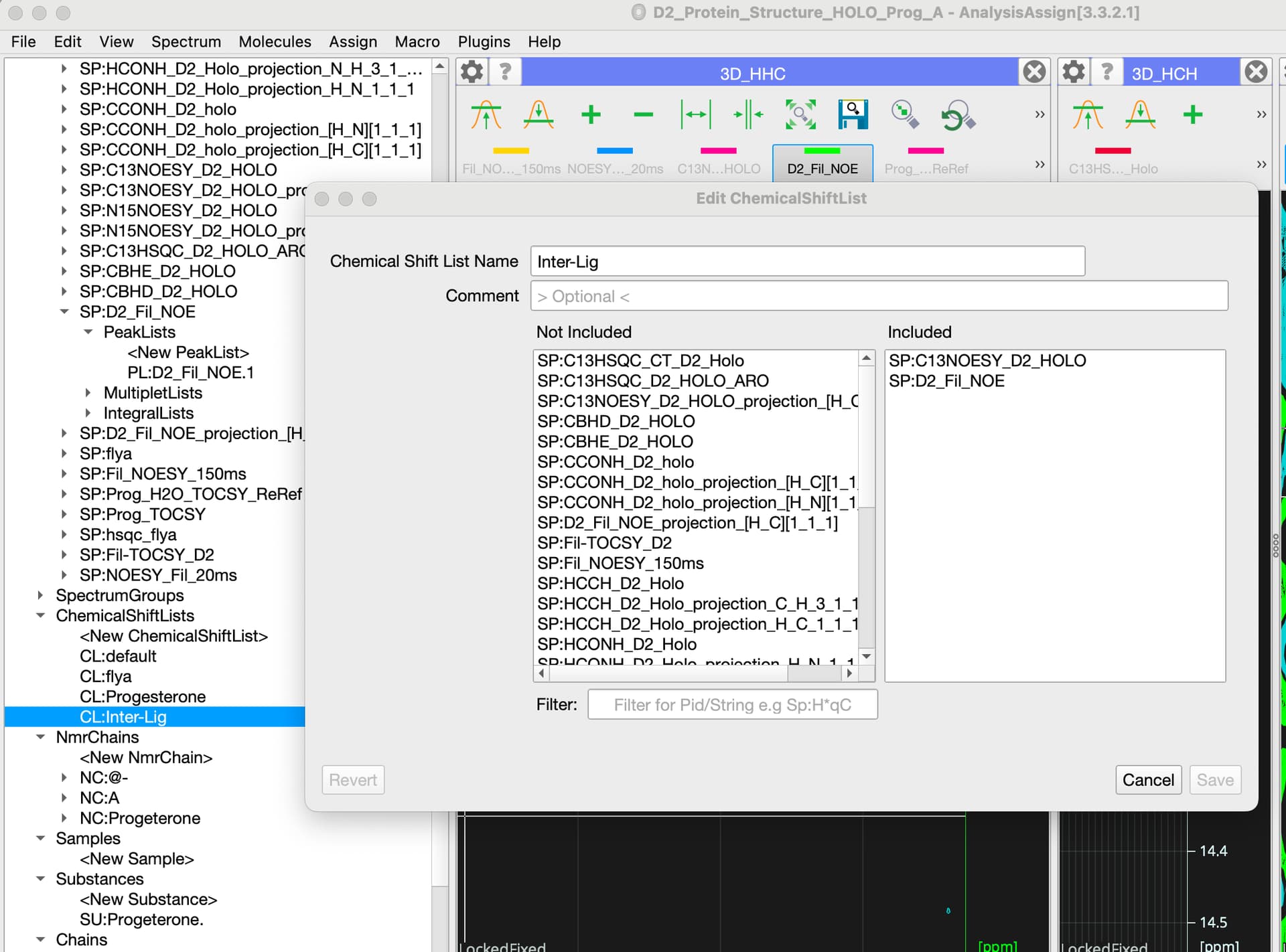
For double filtered experiments you can put spectra in the separate chemical shift list that would only contain your ligand nmrAtoms (which has some consequences if you then want to use assignments in different spectra, there is not easy way to merge those).
This kind of functionality in Peak Assigner would require filtering on the basis of chain and/or type of experiment. This is something that would be great to add in future.
Thank you for bringing this to our attention, I do admit we still have some work to do to support ligand - protein complexes.
BW,
Eliza
Hi Mark,
on the chemical shift list front I would say that everything should be in the same Chemical Shift List: essentially each ChemicalShiftList should be unique to a sample under certain conditions. So for a protein-ligand complex you want all the spectra in the same ChemShiftList, regardless of which molecule you are detecting in the spectrum. But they should be in a different ChemShiftList to your free protein (or the complex at a different temperature/pH etc.).
As Eliza mentions, there are no real filtering tools in the Peak Assigner at the moment. But I think that is the only place where you would really need this information, right? Or is there another place where you would want the program to know about which NmrChain is visible in which spectrum dimension?
Vicky
Hello Eliza and Vicky,
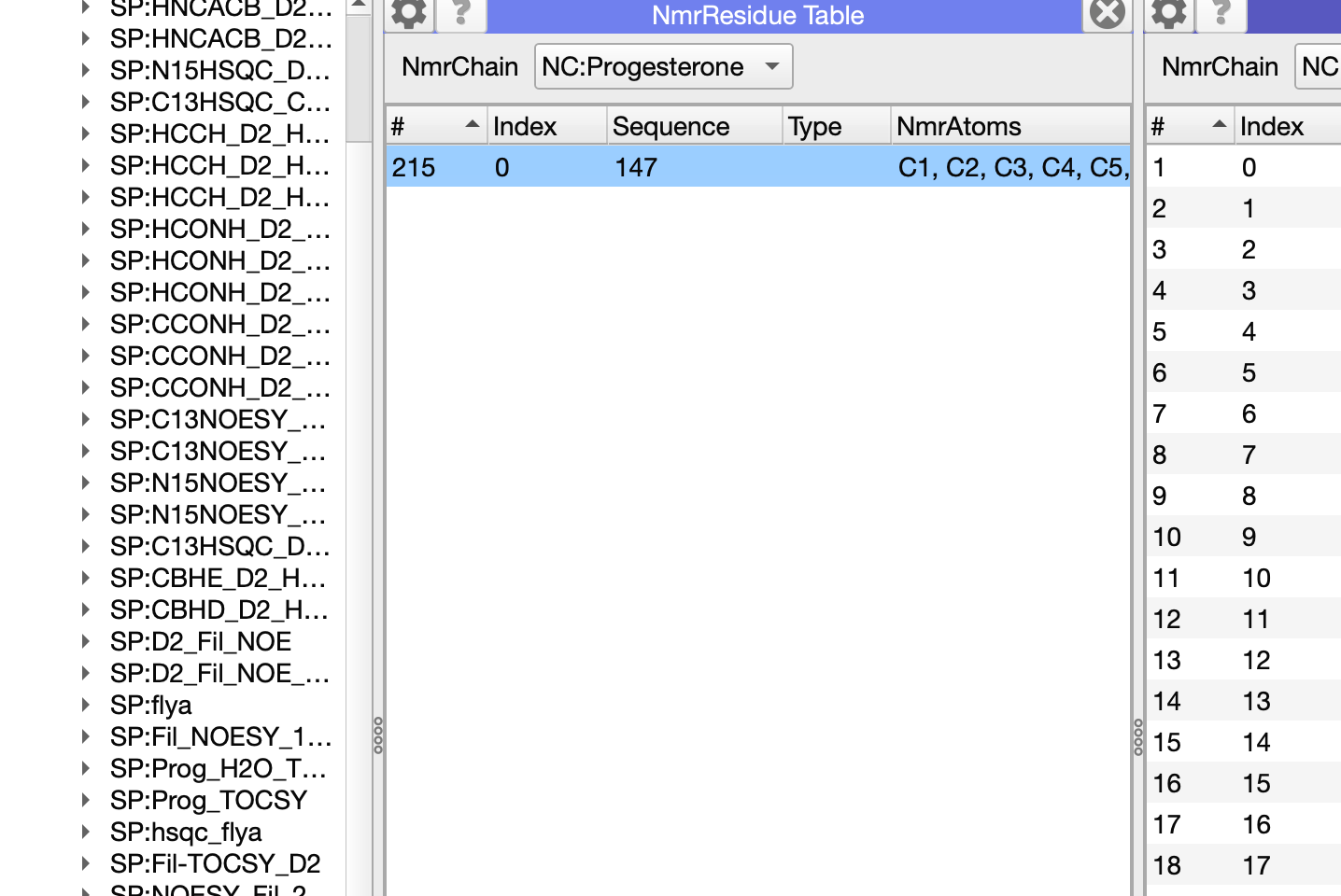
this is very helpful. I spoke to Charles Schwieters as to how to calculate the structure of the complex in Xplor. Since we already have an NMR model for the Holo Protein alone from the ARTINA/NMRtist he suggested a docking protocol (xplor-nih-3.10/eginput/dock_dipolar_chemshift). It seems the ligand binding pocket is likely solvent accessible and we can titrate in the ligand to the Apo protein. In the protocol the ligand is given residue number 147 (protein is 1-117) and two separate molecules are defined for each. Is there a way to make the Sterol ligand residue number 147 in Chain C in CCPN? That would make parsing any lists easier?
Best, Mark
Hi All,
I think I found it? I clicked on the line/residue in the NmrResidue table (displayed NmrChain NC as a module first). Then I was able to change it. Is this correct?
Best,
Mark
Hi Mark,
Thank you for sharing and for the pointer to the Xplor NIH protocol.
When you imported your ligand, did you use the chemComp?
(relevant for ligands as well)
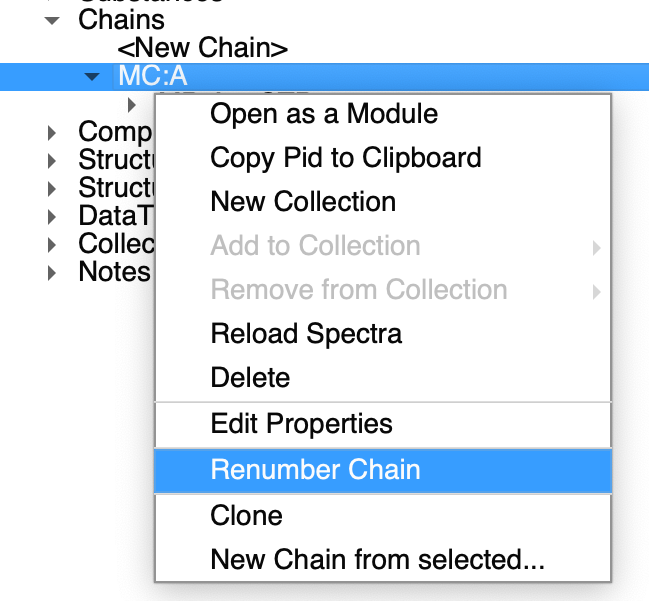
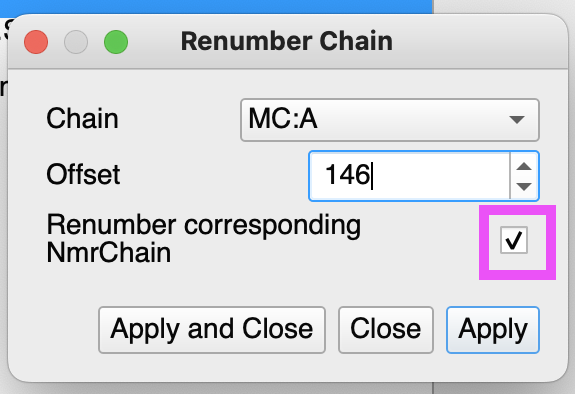
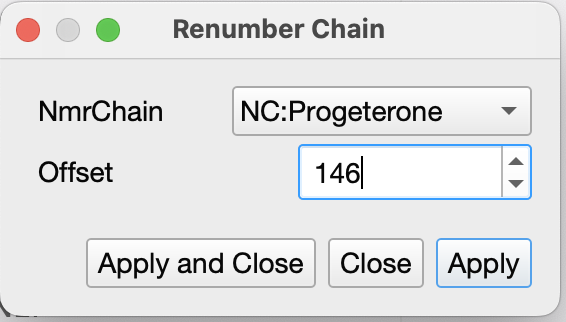
You can renumber chain and consequently nmrChain(residues):
That will make sure both your nmrResidue and molecular chain (that is exported to NEF) has the same numbering.
BW,
Eliza
Hi Eliza,
yes, I imported the ligand I did it via chemComp.
The renumber chain seems to work. My Renumber Chain window is slightly different than yours and is missing the Renumber corresponding NmrChain tickbox, however, it seems to work fine.
I see if you do it this way, you do not need to do edit NmrResidue, and with the offset set to 146 in the Edit NmrChain pop, the ligand get’s assigned residue #147.
Best,
Mark
Hi Eliza and Vicky,
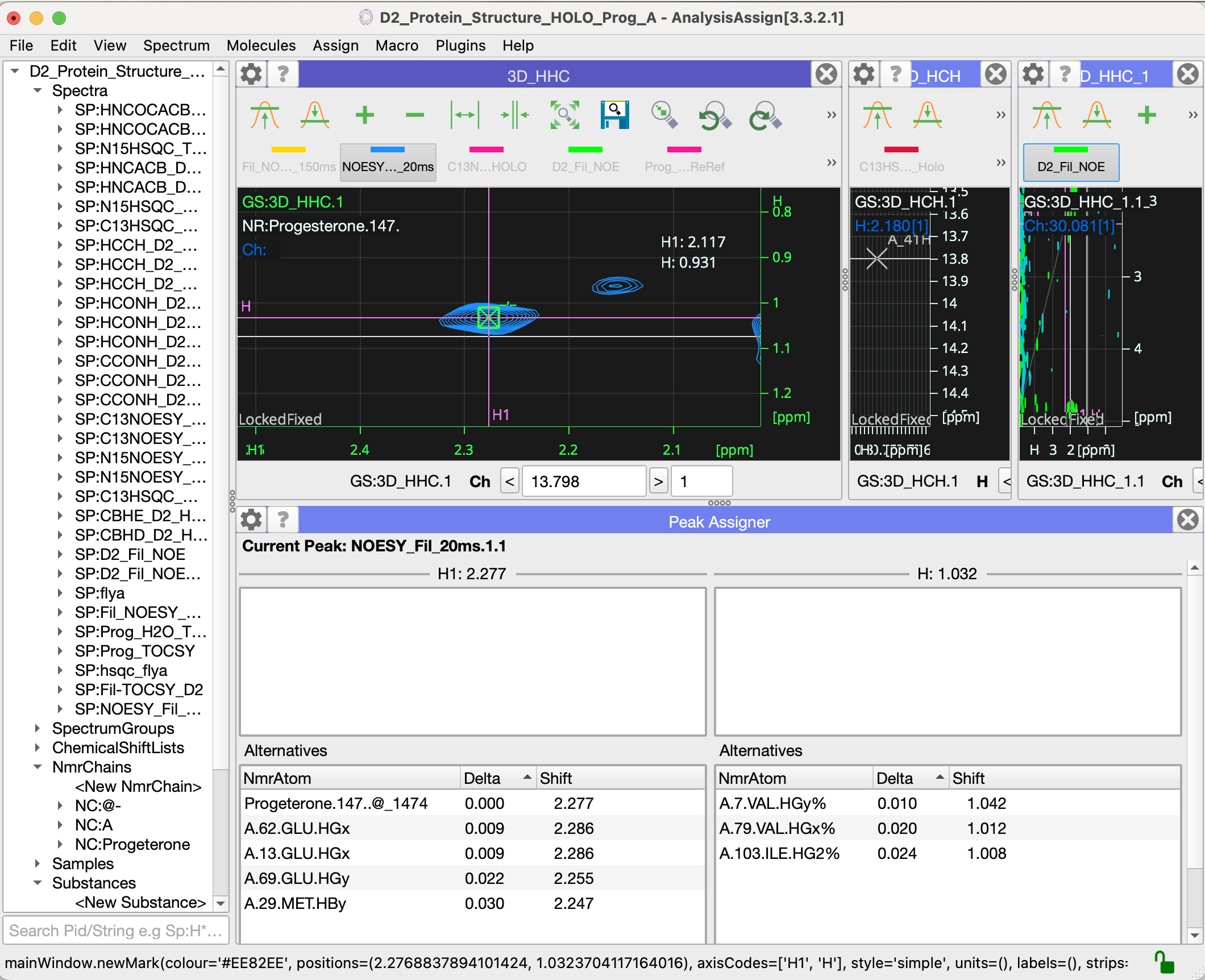
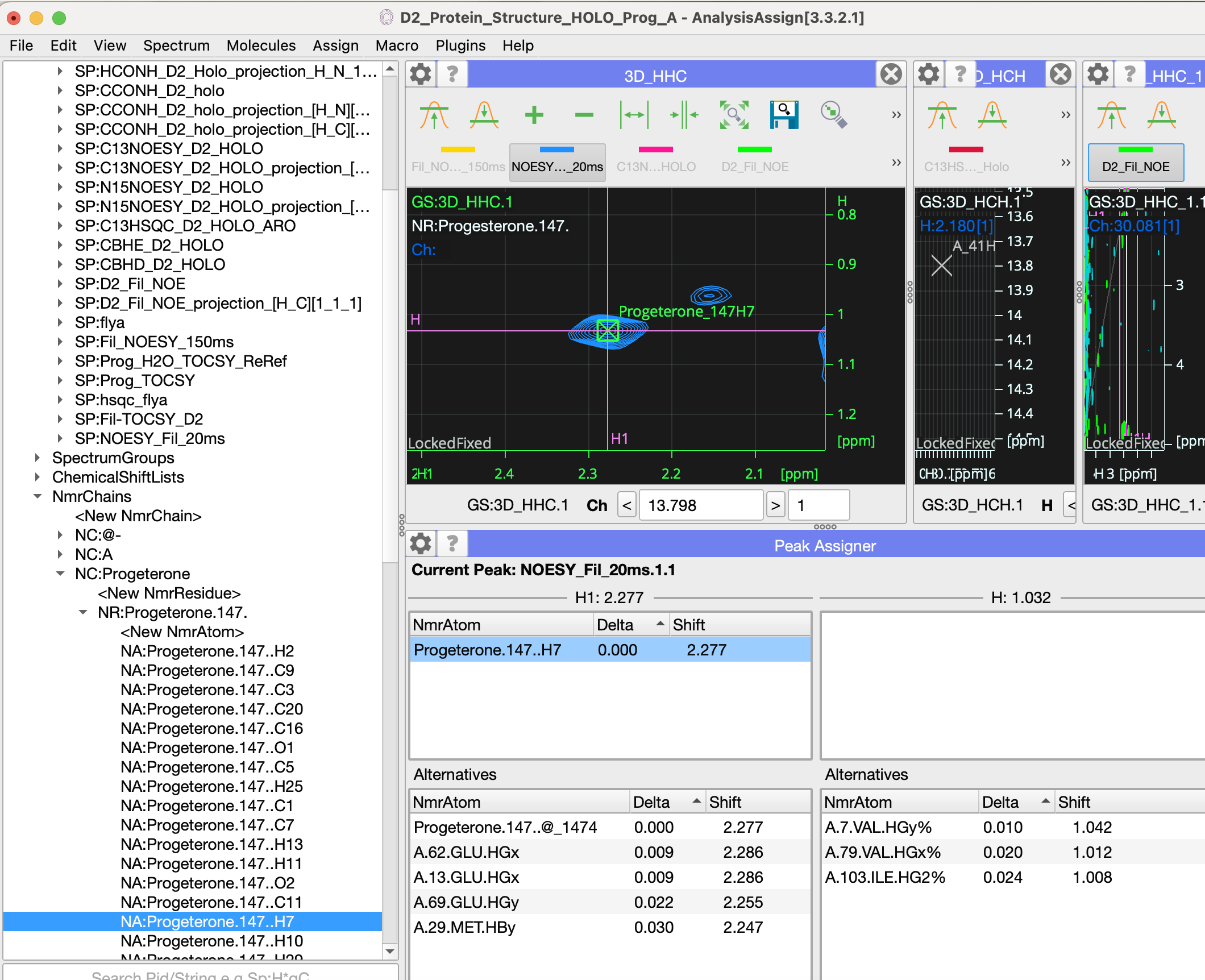
I have, hopefully, just one last question for now: how do I assign ligand resonances to crosspeaks in spectra? When I do edit assignments for the peak below I see the window below. I tried add NmrAtom and also setup NmrResidues, but I wasn’t sure either was the right way to go?
Best,
Mark
Hi Mark,
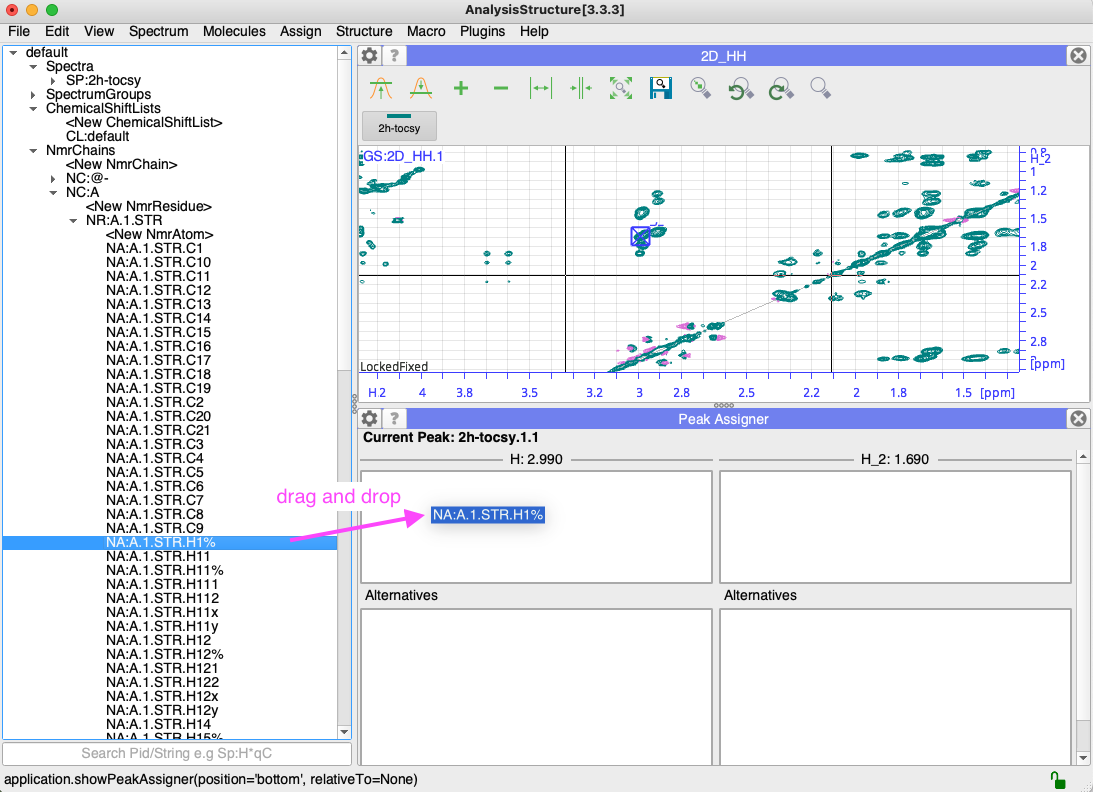
So once you made your nmrChain (and nmrResidue with nmrAtoms) i find it the easiest to drag from the side bar:
Does this help?
If not please let us know!
Eliza
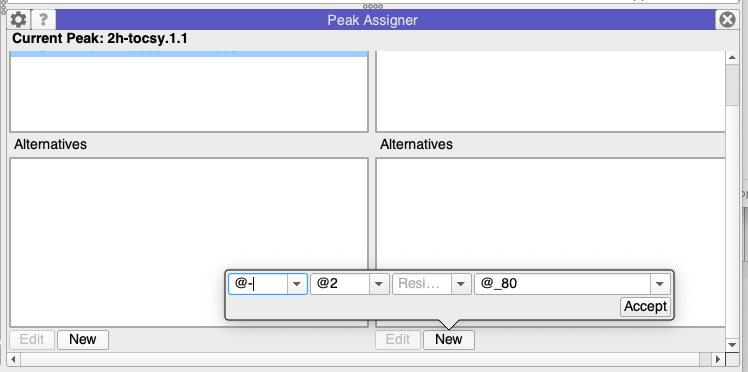
Just to add to this, in principle you should also be able to use this option:
BUT - at the moment this only works for standard residues unfortunately!
It is on the list of things to fix!
Hi Eliza,
Ah, I see. I’m still an L driver with version 3, but I’m slowly getting a better feel for how things are set-up.
What was wonderful, was when I went to read in the structure.nef files from ARTINA/NMRtist, was that I could displays the nOe assignments and peaks, on the 3D NOESY in CCPN. Thanks for getting this importing to work so nicely. It really streamlines checking the assignments and output from ARTINA/NMRtist. It is really helping people here (Students and Postdocs) with transitioning from manual assignments to Ai assisted assignment.
Best,
Mark