It includes the analysis of T1, T2 and heteronuclear NOE data. You can do the R2/R1plot and reduced spectral density mapping, plus generate a some other plots derived from this.
It should all work if you follow the instructions, but isn’t yet as robust as it should be (e.g. you have to stick with s and not use ms for the units!). And I’m sure over time we’ll change a some of layouts etc.
Hi Vicky,
I have problems analyzing my dynamic data. When I created the new spectrum group, on the Series tab, I put the T1 or T2 series values and saved, but these values are not being saved. How can I solve this problem?
Hi Karen, I believe this is a bug on the Popup and the sorting on the first time you create a series. Try to write first your series values as needed as float without clicking sort ascending/descending buttons, then save. Open again the popup and check if the values are stored. Now you can sort the values, if the save button is greyed out, make a small modification on the comment or other entry to renable the save button…
Hi Karen,
For our fitting models we use the package called Lmfit and the default optimiser is the Levenberg-Marquardt or leastsq .
The standard errors for the fitted variables are automatically calculated from the covariance matrix, (the square root of the diagnoal entries of the covariance matrix).
You can find out more about the optimiser and statistical results at Performing Fits and Analyzing Outputs — Non-Linear Least-Squares Minimization and Curve-Fitting for Python
Hello Vicky,
I am getting different results when using this equation in excel vs the results from the chemical shift mapping module. I set the relative contributions like this: alphaH=1 and alphaN=0.1
Not only the absolute values are different but the relative values from residue to residue are different (different bar plot pattern)…could you please have a look at this?
Thanks in advance and best wishes,
Carolina
the best thing would be if you could send me your project (without spectra) to support@ccpn.ac.uk and I could look into this. We’ve actually got a new version of the ChemShift mapping module (accessible under CCPN macros) which hopefully does give the correct results. But we are still in the process of creating the documentation for this.
I’ve had a look at this using our new Chemical Shift mapping module and definitely do get the same results in Excel as in the program. We won’t have the proper documentation for this done until late next week, so in the meantime you might like to look at the following video which will explain how to use the new ChemShiftMapping module and also explains how we calculate the DeltaDelta values.
Exactly what the old CSM module does, I’m not sure. But we are about to retire that, so I would definitely go with the new module which is much more flexible than the old one.
I have diligently attempted to run the RMSD calculation using CCPNv3, but every time I encounter the following message:
“RuntimeError:
Cannot compute the model. Please ensure that the input data contains the T2 experiment.”
I’m confident that all the necessary data is present in my project, and I’m experiencing the same issue in another project with different data. How can I resolve this?
hard to diagnose this without seeing you project. Would you be able to send us one of your projects to support@ccpn.ac.uk (no need to include the spectra)?
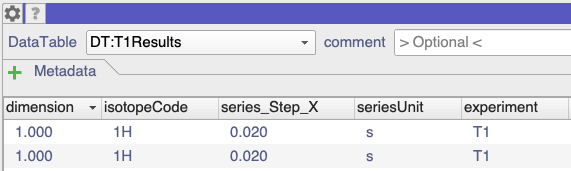
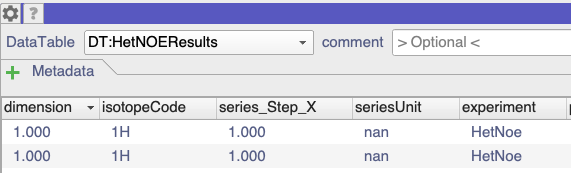
It turns out that your T2 data wasn’t marked as being T2 data. If you look at the DataTables (simply open them as modules by dragging into the drop area from the sidebar), you can see that the T1 and HeteronuclearNOE data was marked as such in the experiment column:
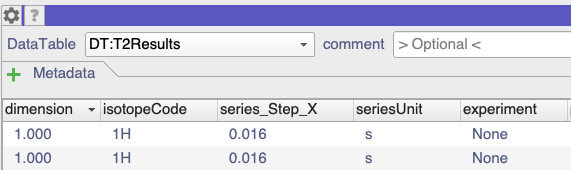
However, the T2Results data simply has None in the experiment column:
I suspect this is because you accidentally had the Experiment Name in the Setup tab set to User-Defined rather than T2.
I was able to correct this very easily with a few commands on the Python Console (open with Space, Space):
import ccpn.framework.lib.experimentAnalysis.SeriesAnalysisVariables as sv
dt = get('DT:T2Results')
dt.data[sv.EXPERIMENT] = 'T2'
And then the R2/R1 calculation and Reduced Spectral Density Mapping work.
We’ll have a think about how we can make it a bit more user friendly to correct such mistakes (as they can easily happen).
Sorry for the delay, I was participating on a congress.
Now everything on my project works well, also I was able to correct other projects by python commands that you have send to me.
CCPN V3 doesn’t have the ability to perform a Lipar-Szabo model free analysis and calculate S2 order parameters at the moment.
There are relatively easy homebrew ways of exporting data from CCPN v3 to use as input for other software (ModelFree, relax, etc.) and we are working on a full pipeline to make this much easier in the coming months.
I also have ‘none’ in the experiment even though in the setup I chose T2. I guess it’s a bug.
I also would like to ask you if you guys have plans to reactivate the ‘backbone’ option in the appearance tab. It was very useful in the past versions.