Hi together.
How can I make Ccon assign recognize that my molecule is an RNA, and show RNA residues instead of amino acids? I would also like to import RNA chemical shift lists and set up dihedral and distance restraints lists, in order to export everything as NEF for deposition. Woiuld be great if someone could help!
thanks
Anna
Hi Anna,
it should be possible to work with RNA in the program, although this isn’t something we have actively supported and tried much. Our main problem (apart from finding the time for it) is that we don’t have any RNA spectra. So if you or someone from your group were willing to share spectra with us and have a conversation about some of your needs, then we can start work on this more quickly and easily.
Now for what there is:
When creating a new chain, you can create both DNA and RNA chains. After double-clicking on in the sidebar you could do the following:
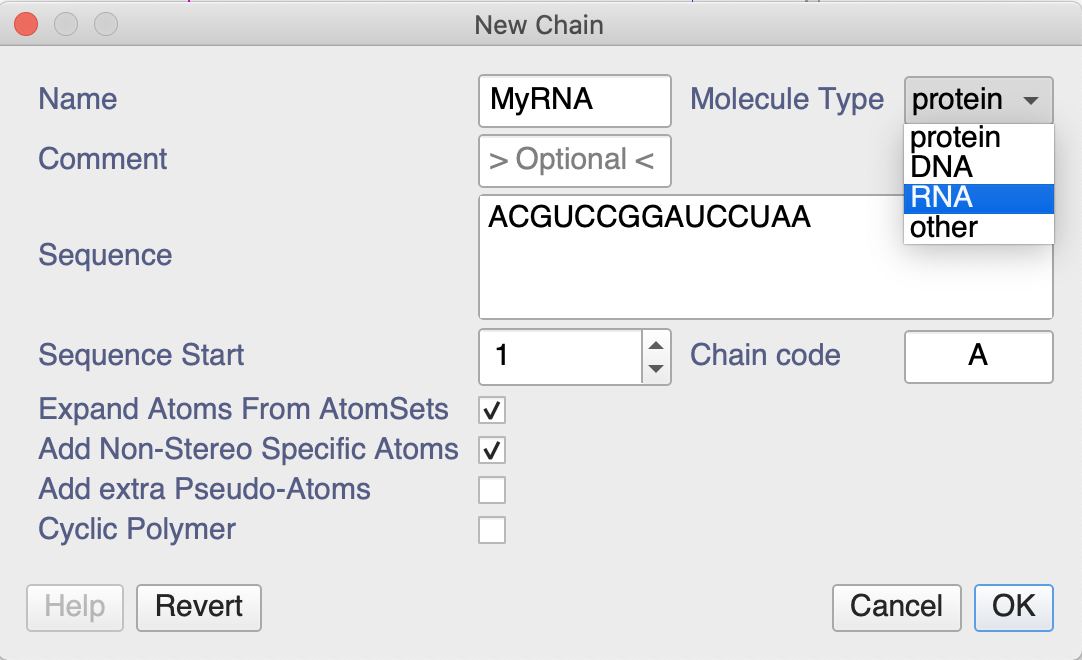
I notice that the drop-down menus, e.g. in the Peak Assigner don’t list the RNA atoms. That’s something we’ll have a look at, though it will have to wait until after our conference and 3.2 release next week. However, you can type whatever you want into the NmrAtom Name box and don’t have to use the drop-down menu. So just type whatever your atom is called.
You can always create an NmrChain from your Chain, if you wanted. After double-clicking on in the sidebar you could do the following:
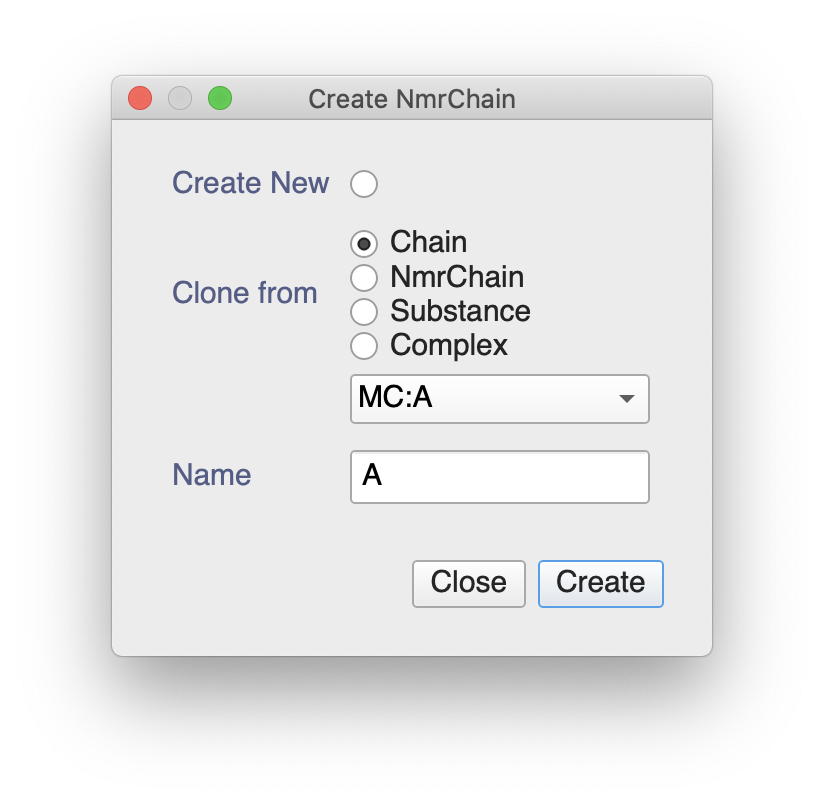
This will give you an NmrChain with all the NmrAtoms in your RNA molecule. You can use these to assign peaks by dragging from the sidebar on to a selected peak.
With regard to importing RNA chemcial shift lists and dihedral restraint lists: it depends a bit on what format your data is in. You can import BMRB and NEF files, but presumably your data are in a different format at the moment?
There are probably several routes you could take at this point:
- change your files into NEF files by hand. Not entirely recommended, as it can be error prone, but we certainly do this from time to time. We have a tutorial (How to create a NEF file form tabular data) which will give you some advice on how to do this at Tutorials - CCPN - Collaborative Computing Project for NMR .
- You could try to import your data into Version 2 using the Format Converter. If you then save your project, you should be able to open it Version 3 and then create the NEF file.
- @varioustoxins has written some software called NEFPipelines which can do some data conversion. We have a tutorial on that, but I don’t know what formats it can currently handle and what it does with RNA data. But I’ll ask him to take a look at this thread and let us know if it might be of any help.
The key thing, however, will be to know what format you currently have your data in.
But do remember: the software is completely flexible about what your NmrAtoms and NmrResidues are called, so although much of the software currently assumes you are working with proteins, you can name things as you like and most of the basic functionality will be fine.
Vicky
Hi Vicky,
amazing how quick your response came! I would be honored to provide you everything I can in terms of RNA spectra, RNA data, and whatever you need in order to work on RNA project :-) And happy to stay in touch anytime. Just let me know the channel.
My restraints file are currently in ARIA format, as I did structure calculations with an in-house-adapted version of ARIA CNS with peak lists and chemical shift lists imported from Sparky. And now I am running into problems with depositing my structure… From everything I gathered around regarding efficient NMR-data-structure-pipelines, the solutions and workflows offered by Ccpnmr sound most convincing to me.
So, as I said… Happy to contribute!
Best wishes,
Anna!
Hi Anna
NEF-Pipelines has facilities for reading sparky sequences, shifts and peaks and has been tested on RNA / DNA for these formats. Also let me know what the format of the restraints you want to read from Aria and I maybe able to help
regards
Gary
links to nef piplelines installation info
via pip nef-pipelines · PyPI
via pipx GitHub - varioustoxins/NEF-Pipelines: Nef tools
nef pipe-lines tutorial [with instructions on how to install NEF-Pipelines if you have already installed ccpnmr]
Also please note NEF-Piplelines is new software there maybe bug, so please don’t hesitate to come back to me if you need advice, get stuck or find problems…
Gary
Hi Anna,
that would be super, if you were able to give us some data - many thanks! Probably best if you contact us by e-mail at support@ccpn.ac.uk and then once our conference (and possibly also EUROMAR) is over, we can be in touch about all of that.
Given what Gary has written, it looks as though NEF Pipelines might be you best bet for the data conversion. Do take a look at the NEF Pipelines Tutorial at Tutorials - CCPN - Collaborative Computing Project for NMR to find out how to install this - the code is included in the latest version of CcpNmr Analysis and I found it fairly straight forward to install and update.
The development version of ARIA is NEF compatible (and @bardiaux is usually happy to give this people who request it from him) - but that may not work well with your in-house adapted version.
Vicky
Hi Gary,
thanks for reacting so quickly to my questions! True, I have seen this possibilities in the NEF pipelines, but I had no clue how to include all the restraints file into one single NEF. I haven’t installed that yet, is it also running on the NMRbox? Otherwise, I will have a try and let you know at which point I’ll fail… ;-)
Thanks a lot again,
best regards
Anna!
alright, thanks again a million times for your advice and I hope you’ll have a successful Euromar!
Will contact your support account in a few days.
best regards,
Anna!
Hi Anna
I don’t think its on nmrbox yet, sorry…
regards
gary
Hi Anna
let me know what you have in the way of restraints files and I can have a look. You can always send an example of them to my kent address
Regards
Gary
Hi Anna
thank you for sending me some files to try out. I have updated nef pipelines to version 0.1.44 which improves support for RNA/DNA and reading of CNS restraints files (use the xplor input distances tool). Note it may take a few minutes to for 0.1.44 to appear…
here is an example command for a couple of you files and its easy to extend it for the rest. Please note that xplor input distances can’t read complex restraints including or statements but should work for you noe tables and h bond restraints
nef fasta import sequence --molecule-type rna --starts 6 seq.txt \
| nef xplor import distances NOE1.tbl \
| nef sparky import peaks --molecule-type rna NOE1Goncalo_298_D2O_NOESY_150_new800.list
notes
- xplor rdcs are not currently supported but is on its way
- sparky peaks doesn’t currently support ran but will do soon
- import of sparky currently requires the flag --molecule-type rna but this will disappear soon
- you need to specify the correct start of the rna sequence otherwise the tools will exit with some rather cryptic errors!
0.1.44 is now released
also please do check the results match what you expect, much of NEF-Piplines is new and there can be bugs…
ps I have also checked import of xplor dihedral restraints and this is also appears to be working but currently is not reading the input nef stream correctly (it forgets any previous frames). The workaround is to run it standalone and copy and paste the output. I will fix this soon…!