Hello!!
I want to calculate the secondary structure of my protein after backbone assignment. when i use the CalculateSecondaryCACB.py
macros, i get an error:
AttributeError Traceback (most recent call last)
File ~/NMR/ccpnmr/src/python/ccpn/macros/macro_yeo76y7c.py:104
102 showWarning(‘chemicalShift list not found’, ‘Set the chemicalShiftList pid in the macro.’)
103 df = chemicalshiftList._data
→ 104 filteredDf = df[df[csl.CS_ATOMNAME].isin(filteringAtoms)]
105 resultDf = pd.DataFrame(columns=[csl.CS_SEQUENCECODE, csl.CS_RESIDUETYPE, CA, CB])
106 for ix, row in filteredDf.iterrows():
AttributeError: type object ‘ChemicalShiftList’ has no attribute ‘CS_ATOMNAME’
There is an issue in a core class that compromised the import of the ChemicalShiftList module, therefore cannot find the namespaces.
I will correct that on the main release. Meanwhile, you can change your macro as following:
replace:
import ccpn.core.ChemicalShiftList as csl
with:
from ccpn.core.ChemicalShiftList import *
remove all the
csl.
instances.
So line 104 will be like:
filteredDf = df[df[CS_ATOMNAME].isin(filteringAtoms)]
107 like:
residueType = row[CS_RESIDUETYPE]
etc.
Unfortunately I don’t think Verison 3 currently or ever will doesn’t support dangle… sorry. I think you would need to export NEF from V3 go back to version 2 and run dangle there on the imported NEF. Of course you could run TALOS and there is support for doing that via NEF and NEF-pipelines…
regards
Dr Gary S Thompson NMR Facility Manager
CCPN CoI & Working Group Member
Wellcome Trust Biomolecular NMR Facility
School of Biosciences, Division of Natural Sciences
University of Kent, Canterbury, Kent, England, CT2 7NZ
Dr Gary S Thompson NMR Facility Manager
CCPN CoI & Working Group Member
Wellcome Trust Biomolecular NMR Facility
School of Biosciences, Division of Natural Sciences
University of Kent, Canterbury, Kent, England, CT2 7NZ
Hi Slava,
It seems you just need chemical shift and chain to run Dangle, to avoid potential errors I would just export that and untick ‘ccpn’ tags, as you do not need those.
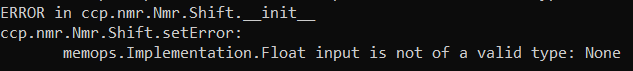
Also you may want to duplicate chemical shift list first and tidy up the new one - maybe delete any shift that you will not need and may cause a problem when importing to V2.
Sorry, should have noticed that earlier, this is because V2 require value error in shifts, and we changed how this is handled in V3. The easiest is just to edit nef file in the text editor and replace ‘.’ with 0.0